|
Ciljano liječenje, stanični mikrookoliš i tumorska distribucija u kroničnoj limfocitnoj leukemiji (B-KLL) |
HRZZ IP-2018-01-3453 |
|
Ciljano liječenje, stanični mikrookoliš i tumorska distribucija u kroničnoj limfocitnoj leukemiji (B-KLL) |
HRZZ IP-2018-01-3453 |
|
U nastavku se nalaze hrvatske (KROHEM) smjernice za dijagnozu u liječenje kronične limfocitne leukemije. Smjernice su objavljene u ACC te na stranicama KROHEM-a na kojima se objavljuju amandmani. GUIDELINES FOR DIAGNOSIS AND TREATMENT OF CHRONIC LYMPHOCYTIC LEUKEMIA. KROHEM B-CLL 2017
Branimir Jaksic1 Vlatko Pejsa2 Slobodanka Ostojic-Kolonic1 Ika Kardum-Skelin1 Sandra Basic-Kinda3 Bozena Coha4 Velka Gveric-Krecak5 Radovan Vrhovac3 Ozren Jaksic2 Igor Aurer3 Jasminka Sincic-Petricevic6 Antica Nacinovic-Duletic7 Damir Nemet3 for KROHEM CLL Working Group
1Merkur University Hospital, University of Zagreb Medical School 2Dubrava University Hospital, University of Zagreb Medical School 3Zagreb University Hospital Center, University of Zagreb Medical School 4Slavonski Brod General Hospital 5Sibenik General Hospital 7Osijek University Hospital Center, University of Osijek Medical School 8Rijeka University Hospital Center, University of Rijeka Medical School
Correspondence to: Branimir Jaksic, KROHEM (Croatian Cooperative Group for Hematologic Diseases), Buzinski prilaz 10 HR-10010 Zagreb, Hrvatska; E-mail branimir.jaksic@zg.ht.hr
Summary Recent developments in diagnostics and treatments of chronic lymphocytic leukemia (B-CLL) have led to change of approach in clinical practice. We define a current position of Croatian Cooperative Group for Hematologic Disease in the transition from chemo-immunotherapy paradigm into a new one that is based on new diagnostic stratification and unprecedented therapeutic results of B-cell receptor inhibitors (BRI) and Bcl-2 antagonists. This is rapidly evolving field as a great number of currently performed clinical trials constantly accumulate and provide new knowledge. This is helping to refine concepts and lead to more effective clinical care based on individualized precision medicine.
Key words: Chronic lymphocytic leukemia; gudelines; CLL; Krohem
SAETAK - Nedavni događaji u dijagnostici i liječenju kronične limfocitne leukemije (B-CLL) doveli su do promjene pristupa u kliničkoj praksi. Definiramo trenutni stav Hrvatske kooperativne grupe za hematoloke bolesti u sadanjoj tranziciji iz kemo-imunoterapijske paradigme u novu koja se temelji na novoj dijagnostičkoj slojevitosti i izvrsnim terapijskim rezultatima inhibitora B-staničnih receptora (BRI) i Bcl-2 antagonista. To se područje brzo razvija kako veliki broj kliničkih ispitivanja koja su u tijeku bez prestanka doprinose i pruaju nova znanja. To pomae poboljanju koncepata i vodi do učinkovitije kliničke skrbi na temelju individualizirane i precizne medicine.
Ključne riječi: Kronična limfocitna leukemija; Smjernice; KLL; Krohem
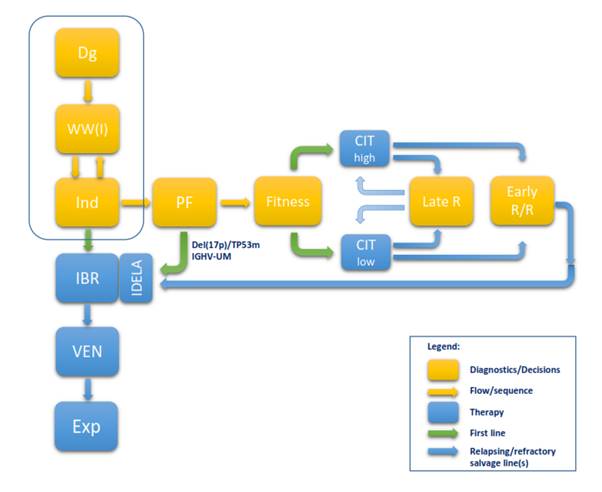
Contents Setting the suspicion and the referral of patients to hematologist. The diagnosis and differential diagnosis Evaluation of the stage / extent of the disease Prognostic factors at diagnosis and predictors of response Indications for treatment / the criteria for initiation of therapy The schedule of activities and assessments Therapy individualization - defining goals and strategies, and stratifying patients Therapeutic recommendations based on evidence / clinical trials Choice of antineoplastic treatment options - new therapeutic paradigm Treatment options and current labels for approved antineoplastic medications: Criteria for evaluation of response to therapy Antineoplastic therapy current treatment options Management of patients with no accepted criteria for therapy Initial treatment (first-line treatment) Treatment for relapsed / refractory CLL Consolidation / Maintenance therapy The role of allogeneic transplantation Myelodisplastic syndrome and acute leukemia Table 2. Clinical stages and TTM Table 3 Criteria for active (progressive / symptomatic) disease Table 4 General schedule of investigations before, during and after therapy Table 5 Definition of response to treatment (NCI updated guidelines, Blood 2008) Figure 1 Flow chart describing 2017 paradigm shift Table 6 FIRST LINE TREATMENT OF CLL (KROHEM v1 2017) Table 7 TREATMENT OF RELAPSED/REFRACTORY CLL (KROHEM v1 2017)
Introduction and definitionIn recent years dramatic change in therapeutic landscape led to unprecendented therapeutic results that translate to clinical practice and after years of slow to modest progress in the field B-chronic lymphocytic leukemia ASCO named transformation od CLL treatment Cancer advance of the year for 2015(1). New tratments were approved based on results of randomized multicentric trials for first line and for salvage already, and the results of numerous ongoing clinical trials are contiuousely providing answers and further refining therapeutic strategies. This is paralled with an explosive growth of undestanding the disease genetics due to major advances in next generation sequencing (NGS) technology(2, 3). All this led to change of until now predominant paradigm based on chemo-imunotherapy (CIT) into a new one(4, 5). Definition. B-cell chronic lymphocytic leukemia (B-CLL) and related disorders (monoclonal B-lymphocytosis-MBL and small lymphocytic lymhoma-SLL) are defined by the presence of clonal mature B-lymphocytes with typical immunophenotype in peripheral blood, bone marrow and lymphoid organs (WHO, iwCLL)(6, 7) representing one nosological entity. Today it is considered that these entities are different manifestations of the same disease. MBL is the most prevalent and is considered an early stage of malignancy that in about 1-2% per year progress to CLL/SLL. SLL represnts less than 10% of overt malignancy, and for this reason B-CLL is most commonly used to to represent both variants (CLL/SLL)(6, 8-17). Epidemiology. B-cell chronic lymphocytic leukemia (B-CLL) is the most common type of leukemia in Western countries, the incidence is estimated to more than 6 per 100,000 people annually. The median age at diagnosis is growing globally, so that now exceeds 70 years. It should be noted that the age at start of treatment is several years more than age at diagnosis, depending on duration of observation without treatment. The disease is nearly twice as common in men(18-22). The incidence and prevalence of MBL is much higher, depends on sensitivity of the methods used and is estimated to be up to 12% in population aged over 40 years (23, 24). Diagnostic procedureThe diagnostic process can be conditionally divided into several sections (steps, phases) with respect to the different objectives to be achieved. In Table 1., the basis for the decision is specified, the main criteria for classification and possible categories to which the classification in this section should lead. Setting the suspicion and the referral of patients to hematologist.Today, the most frequent finding leading to suspicion to B-CLL is absolute lymphocytosis during routine blood examination (70-80%), and less frequently (20-30%) is due to the finding of organomegaly (swollen lymph nodes and/or spleen) or symptoms associated with CLL. The diagnosis and differential diagnosisThe activities that follow are directed toward defining the cell type, enabling the diagnosis (type of disease) and differential diagnosis. It is mandatory to make the diagnosis of a typical B-CLL on the basis of morphology and flow cytometry in the peripheral blood sample and to distinguish it from other entities in the CLL syndrome. For the diagnosis of B-CLL phenotype (typical phenotype) the following is required: The restriction of sIg light-chain expression of low intensity, CD5+, CD19+, CD20low, CD23+ (25). Basic hematological clinical findings & blood count allow quantification of the tumor mass in the peripheral blood and lymphoid organs, which allows the classification of entities that meet the diagnostic criteria for: (1) B-CLL (presence in the blood of more than 5 x 109 / L clonal cells) or (2) SLL (less then 5 x 109 / L clonal cells in blood and present clonal lymphadenopathy greater than 1.5 cm, and (3) MBL (less then 5 x 109 / L clonal cells in blood and no clonal lymphadenopathy or symptoms. So, a different size of the tumor mass between B-CLL and MBL is critical and the tumor distrbution is critical for distinction between B-CLL and SLL. It is evident that for the diagnosis of type of disease, a very small number of tests is enough, because if the result is positive in the peripheral blood (PB) it is not necessary analyze the bone marrow (BM) or lymph nodes (LN), although these tests have their place in further diagnostic procedures. For the diagnosis of SLL it is recommended to do node biopsy to establish the diagnosis and to distinguish between MBL and SLL the radiologic assesment (US or CT) of neck, thorax, abdomen and pelvis can be useful. Differential diagnosis encompas other entities that may have increased counts of lymphoid cells in blood. The distinction to CLL is usually made on flow cytometry assessed imune phenotype. Majority are B-cell malignancies like follicular lymphoma (FL), mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), prolymphocytic leukemia (PLL) and hairy cell leukemia (HCL) that all together represent less than 15% of all lymphoid appearing leukocitoses, and less than 5% expres T-cell lineage markers variants. It should be remebered that this type of disorder classification based only on the immune phenotype is not using morphological, cytogenetic, nor molecular or other characteristics of the diseases that today show significant association with prognosis. Evaluation of the stage / extent of the diseaseAfter the diagnosis of the type of disease, the evaluation of disease stage or extent follows. This is in principle is made on the basis of clinical and hematological evaluation, under the criteria set out in Table 2. Clinical stages represent a simple tool for the clinical assessment of the disease extent. The basis of these systems is the assumption that the disease is gradually progressing and expanding. Therefore, the patients who have advanced disease have a higher tumor load and more extensive disease. (26-28) The clinical stages according to Rai and Binet assess tumor size by simple parameter estimates of the size of the tumor mass (without precise quantification of the affected comparments) along with the parameters for the assessment of bone marrow failure. In doing so, the greatest prognostic power contribution has the failure of the bone marrow. Note that it is not a direct sign of tumor size but an indirect one. The prognostic power is relatively weak if the failure of the bone marrow is excluded. Estimate of the size of the tumor mass (TTM) is different in that it quantitatively evaluates the tumor mass in 3 major cell compartments, regardless of bone marrow failure (Table 2). Quantitative character enables unbiased monitoring of disease progression and is a very convenient tool for the assessment of therapeutic response (see later). In addition, in most patients who have affected both peripheral blood and lymphoid organs it is possible to determine the type of distribution of the tumor mass by comparing leukemic (TM1) and lymphoid organ (TM2 +TM3) compartments. (29) This features of TTM system enables to estimate the dynamics of disease, progression and response to therapy and redistribution (see later)(30). Prognostic factors at diagnosis and predictors of responseAnalyses of prognostic factors performed in the era when therapy had little effect on clinical course identified a number of clinical or laboratory factors(31) decribing broadly the natural course of the disease. Prognostic factors that can be determined immediately at diagnosis are primarily related to the additional characterization of tumor itself. Some of them do not change during the evolution of the disease such as for example the mutational status of IgVH genes that generally discriminate between more benign and more malignat disease(32-34) and is currently sugested to become minimal standard initial evaluation(31). There were until now rarely used in routine clinical practice and other tests that have a high correlation with mutational status are used instead, such as CD38 and ZAP-70 by flow cytometry and immunohistochemistry, with the higher value was found to correlate with poorer prognosis.(35-45) The usefulness of there surogate factors is still controversial. In contrast, cytogenetics and molecular tests (FISH) to determine: del (11q22-23), +12; del (13q14), del (17p13)(46) showed a strong independent prognostic effect and usually change (progress) over time. Also, finding of certain mutations (including TP53, SF3B1, NOTCH1, etc.)(47, 48) can complement these findings. These tests are good predictors of response in chemo-immunotherapy era and have proven to be useful to stratify patients into groups that react differently to certain drugs(4, 49). Since they may change during disease course, the testing is mandatory performed just before each new line of therapy. All this tests that further characterize neoplastic clone cells are technically demanding and expensive, but relatively inexpensive compared to drugs, and for those validated justify the cost end effort. Predictive factors seem to change or even lose their predictive power with novel treatments. Likewise, failure to the new treatmens will require discovery of new predictors. On the other hand, a number of additional factors more related to patients state also have a strong prognostic power, so a number of different parameters is often used, and multivariate analyzes study their impact on prognosis(50). Because of the above, we distinguish three types of factors, given the causal relationshipwith B-CLL. First, those who are associated with B-CLL clonal neoplastic diseases (size distribution, the growth rate of tumors, mutational status, CD38, ZAP-70, FISH, and may be used as predictors of response to therapy, etc.) (28, 32, 33, 46, 51-53) Also, numerous other characteristcs such as blood chemistry (serum beta2 microglobulin, sCD23 and serum thymidin kinase(54-56), clonogenic(57), kinetic, and computer asissted cell imaging have been shown to be related to prognosis(58, 59); second , those who are associated with organ failure (mixed groups, they may be due to both the very undelying neoplastic diseases and to consequences of associated diseases, such as anemia, thrombocytopenia, whose ethiology should be carefully evaluated); and third, those factors that are associated with the patient but not directly with neoplasm (age, gender, performance status, comorbidity), that correlate with fitnes and the ability to tolerate agressive treatments(40, 60-63). On the basis of strong independent predictors composite prognostic indices calculated(64-66), and most recently CLL International prognostic index (IPI)(67). They show very good prognostic power with respect to the length of survival in chemo-immuno therapy (CIT) era, but musch less so with new treatments. Also, the stratification of patients for treatment that is adapted to risk one should be careful, because the composite index usually consist of the factors that belong to each of the above mentioned three groups. With new, more effective therapies prognostic landscape changes. Most of important predictors in the CIT era lose their power, and new once are still to be identified(4, 49). Indications for treatment / the criteria for initiation of therapyThe criteria for active disease that warrants start of treatment are based on iwCLL criteria, and are amended with TTM (Table 3.). The decision should be based solely on an assessment of the parameters that are associated with neoplastic B-CLL clone(6). For parameters that may be unrelated to the neoplasm, it is necessary to carefully evaluate the extent to which parameters are associated with neoplastic clone (eg fever, anemia, etc.). Criterion parameters can be classified into three distinct types: 1. Quantitative parameters for which is defined threshold consensus that is considered to justify the initiation of treatment, such as anemia, defined by a certain level of hemoglobin, thrombocytopenia, defined by platelet count, organomegaly defined by size of the spleen or lymph nodes. 2. Monitoring data to enable assessment of trends, eg progressive cytopenia, progressive lymphadenopathy and / or splenomegaly, increased leukocyte count or TTM values (see the previous section). It should be emphasized that only the measurement of dynamic parameters can directly evaluate progression of disease, as opposed to the a priori estimate of a possible evolution described in the previous section (see paragraph 2.4 Prognostic features at diagnosis). Here, however, we should point out certain difficulties and ambiguities in quantitative measurement of the dynamic parameters, especially in the early stages of the disease. This imprecision in a priori definition of the criteria for progressiveness, despite a very attractive concept, often leads to delay a decision until the moment when it reaches the absolute value of the threshold that is set up as described under number one. 3. Qualitative criteria of the occurrence of symptoms, threatening organ damage and the like, and which is considered to be the result of neoplastic disease activity. Today, we seek to combine threshold criteria with dynamic criteria, as shown in Table 7. Observe that a number of criteria, the criteria of threshold, dynamics and qualitative changes may be used. Although it is sufficient to indicate treatment by the presence of only one of the stipulated criteria, it is important that this criterion is compelling, and the presence of multiple criteria makes decision certainly easier. For dynamic criteria it is especially important, so it is good to compare the growth trend of the tumor mass with trends of deepening anemia and/or thrombocytopenia. The indication for treatment (according to KROHEM guidance) should be documented in patient records. The schedule of activities and assessmentsTable 4. shows the general scheme of tests that are used in
pretreatment work-up, in therapy monitoring and after therapy. In about 10% of
diagnosed patients indications for therapy are present, so the pre-treatment
work-up is done immediately at diagnosis and visits 1-3 are completed without
delay. Other patients (90% of all diagnosed) are observed after diagnosis at
periodic visits until meeting the criteria for initiation of treatment. At that
point the pre-tretment complete work-up is performed. It is possible to distinguish several specific clinical questions that need to be answered in stipulated visits that require different extent of investigations. These questions are answered in following visits: (1) Diagnosis, differential diagnosis, evaluation of the extent of disease, and preferably but not mandatory assessment of prognostic groups that require investigations (a-f). At this time it is reccommended to evaluate general health profile (j); (2) repetitive visits monitoring of the clinical and hematological parameters that serve as a criteria for initiation of treatment are based on simple parameters (investigations a,b,c,j). The time between visits may vary from weeks to months; (3) Immediate pre-therapeutical work-up provides a definite classification of the disease, a definitive assessment of prognostic parameters as well as the general state of the patient, and any associated illnesses including infection status. For this reason, this visit should most complete, enabling stratification and providing base-line parameters for treatment; (4) these repetitive visits that monitor the course of therapy should be tailored according the needs of respective therapy; (5) end of therapy (EOT) visit should enable evaluation of achieved response. After that, for CIT that is time limited, monitoring visits without therapy (2 ') are repeted again and in case of the need for a new line of therapy repeat visits 3', 4 and 5. The situation is different for novel agents since this treatment is at present time ulimited and visits 4 should enable detection of treatment failure. Therapy individualization - defining goals and strategies, and stratifying patientsAt the end of diagnostic procedure described in sections 1-6, there is a need to make a decision about therapy which wil reflect individualization of given patient. Decision is based on the integration of the factors related to the neoplasm on the one side, with the factors related to the patient on the other side. According to new circumstances and new emerging paradigm because of the lack of curative treatment initial therapy should maximize efficacy while minimize overall toxicity. The overall approach is essentially based on clinical judgement and the expertize In general, we are comparing the two risks: (1) the risk of disease and (2) the risk of treatment. It is clear that the risk of treatment should reasonable lower than the risk of the disease. The risk of disease. For some treatments was shown that are inefficient in some disease subsets. However, novel treatments are also active in those subsets, which will diminish the importance of stratification according to FISH and TP53 which was necessary until now. Neverthless, since CIT proved very effective in some subsets of patients and contraindicated in others, the classification according to risk should remain until head to head comparisons resolve pending questions. The risk of the therapy, i.e. tolerance (or acute treatment toxicity) is highly dependent on the general condition of the patient and the presence of associated diseases, which is often associated (though not exclusively) with the patient's age. However, new B-cell receptor inhibitors (BRI) treatments have much better toxicity profile which diminish the importance of stratification according to fitness which was necessary in chemo-immunotherapy based paradigm. Neverthless, for the same reason as described above this stratification should remain for CIT. Since CIT is not indicated in patients with del(17p)/TP53 mutation, the distinction between fit and unfit become irrelevant. A patient is classified as capable (fit, Go Go), when there is a low comorbidity score (eg CIRS-G <6). Although age does not enter into the calculation for CIRS-G, it is known that age is a very important factor, and it should be taken into account. It is common to impose an age limit for stratification in therapeutic groups. Today the limit is set at 65 years. Patient aged over 65 years can be considered capable for receiving aggressive therapy, if is in excellent health, without substantial comorbidity. On the basis of the two above-mentioned principles patients are today stratified into three strata with respect to antineoplastic therapy: (1) without the deletion of the p53 gene with a good general condition (capable for aggressive, fludarabin based CIT therapy) - (LOW RISK + FIT), (2) without the p53 gene deletions with poor general condition (incapable for aggressive therapy) (LOW RISK + UNFIT), (3) the deletion of (17p) / TP 53 mutation (HIGH RISK + FIT/UNFIT). For those patients no further division according to fitness is not longer necessary, since new approved treatments are well tolerated(16, 17, 68, 69). In other words, these strata may in principle represent the combination of risk (high correlation with TP53 abnormality) and age (high correlation with fitnes). However, for the reasons stipulated above the chronological age cut-off should not be rigid, to allow elderly patients in good health to enjoy the benefit of more agressive treatment and vice versa spare younger patients with co-morbidity of unwarranted therapy associated risks. The stratification is used for guideliness and reflect general principles, but for each patient, the treatment plan should individualized and set-up after careful clinical evaluation taking also into account patients' preferences. Therapeutic procedureTherapeutic recommendations based on evidence / clinical trialsThe treatment for B-CLL consists of antineoplastic therapy and supportive measures. Antineoplastic measures consist traditionally of chemotherapy, therapeutic antibodies, radiotherapy, stem cell transplantation methods and recently novel agents that include B-cell receptor signal transduction inhibitors (BRI) and bcl-2 antagonists. In this gudeliness the reccommendations are based on phase 3 clinical trials, in some cases on evidence from earlier phase trials, and on the approved agents and therapies in Croatia. Combination therapy is generally more efficient then monotherapy. Choice of antineoplastic treatment options - new therapeutic paradigmBecause this disease is generally not curable, occurs in an elderly population, and often progresses slowly, it is most often treated in a conservative fashion. In asymptomatic patients, treatment may be deferred until the patient becomes symptomatic as the disease progresses(70). This concept is still valid and the treatment outside clinical trials is recommended only for patients who fulfil described indications. Since the rate of progression may vary from patient to patient, with long periods of stability and sometimes spontaneous regressions, frequent and careful observation is required to monitor the clinical course(5). At present about 30% live without symptoms and never need therapy. Others sooner or later progress and fulfil the criteria for therapy. The new, very effective treatments recently dramatically changed the therapeutic landscape and led to change in chemo-immuno therapy (CIT) based current paradigm into a new one. In comparison to until now available therapies, the new treatments show markedly improved efficacy and considerably better tolerance. This has impact on all aspects of patients management and choice of treatment options. This goes for the deferring therapy also, but at present there are no yet data to support early treatment. Current and future clinical trials that include novel agents in this setting may change this current concept. For patients in whom criteria for start the therapy are met, the general principle in the new paradigm is that (because no curative therapy has yet been found) initial therapy should maximize efficacy (with improvement of OS), while introducing the least overall toxicity, both short term and long term. Standard chemotherautic agents induce not only cytopenias and sometimes fatal infections (acute treatment toxicity) but also induce mutational damage to the genome that can manifest as more aggressive and refractory phenotypes upon relapse and can induce second malignancies. For this reason avoiding chemotherapeutic agents upfront, when possible, is a new paradigm of sequencing therapy for CLL(5). Major changes are explained in Figure 1. However, in the absence of head to head randomized trials to assess efficacy/tolerance ratio between novel therapies and best CITs for fit patients, for this subset CIT should not be abandoned. Also, the access and availability of new treatments will need some time, and the current cost of novel treatment may be out of reach for insurers. Treatment options and current labels for approved antineoplastic medications:Observation. For patients who do not fulfill criteria for star therapy, the treatment is observation. Althoough this is in contrast to general oncological tendency to treat the patients with neoplasms as soon as possible, no data exist to suggest any harm in deferring the therapy in those patients. Since the rate of progression may vary, frequent and careful observation is required to monitor the clinical course. Ibrutinib. Ibrutinib is a selective irreversible inhibitor of Bruton tyrosine kinase, a signaling molecule located upstream in the B-cell receptor-signaling cascade. Label: IMBRUVICA as a single agent is indicated for the treatment of adult patients with previously untreated chronic lymphocytic leukaemia, IMBRUVICA as a single agent or in combination with bendamustine and rituximab (BR) is indicated for the treatment of adult patients with CLL who have received at least one prior therapy(71). Those indications were approved based on randomized, phase 3 studies RESONATE-2 (PCYC-1115-CA), RESONATE and HELIOS (72-74). Idelalisib. Idelalisib is an oral inhibitor of the delta isoform of the phosphatidylinositol 3-kinase, which is located in the B-cell receptor-signaling cascade. Label: ZYDELIG is indicated in combination with an anti-CD20 monoclonal antibody (rituximab or ofatumumab) for the treatment of adult patients with chronic lymphocytic leukaemia (CLL) who have received at least one prior therapy, or as first line treatment in the presence of 17p deletion or TP53 mutation in patients who are not eligible for any other therapies(75). Approval is based on randomized, double blind, phase 3 study (76) and in phase 2 study (77). Rituximab. Rituximab is a murine anti-CD20 monoclonal antibody. Label: MABTHERA is indicated in combination with chemotherapy for the treatment of patients with previously untreated and relapsed/refractory CLL(78). Approval was based on randomized phase 3 studies with: fludarabine and cyclophosphamide (FCR)(79, 80); Bendamustin (BR) (81, 82); and chlorambucil(83, 84). Obinutuzumab. Obinutuzumab is a human anti-CD20 monoclonal antibody. Label: GAZYVARO in combination with chlorambucil is indicated for the treatment of adult patients with previously untreated chronic lymphocytic leukaemia (CLL) and with comorbidities making them unsuitable for full-dose fludarabine based therapy(85). Approval is based on a randomized phase 3 study(83). Ofatumumab. Ofatumumab is a human anti-CD20 monoclonal antibody. Label: ARZERRA is used in previously untreated patients who cannot be treated with fludarabine; in these patients it is used together with chlorambucil or bendamustine (other cancer medicines); in patients whose disease has not responded to previous treatment (known as refractory disease) with fludarabine and a medicine called alemtuzumab; in patients whose disease has come back after previous treatment (known as relapsed disease). In these patients Arzerra is used together with fludarabine and cyclophosphamide(86). Approval is based on randomize phase 3 study in combination with chlorambucil COMPLEMENT-1(87). Alemtuzumab. Alemtuzumab, the monoclonal antibody directed at CD52. The drug was withdrawn for CLL indication by the producer because of commercial reasons. However, the company can offer on request the drug for compassionate use free of charge. Label: (EMA EPAR 2011 authorization withdrawn)) MABCAMPATH is used to treat patients with B-cell chronic lymphocytic leukemia for whom treatment combinations including fludarabine are not appropriate(88). Approval was based on randomized clinical trials and showed activity in TP53 mutation (89-91). Venetoclax. Venetoclax is a highly selective inhibitor of BCL2. Label: VENCLYXTO monotherapy is indicated for the treatment of chronic lymphocytic leukaemia (CLL) in the presence of 17p deletion or TP53 mutation in adult patients who are unsuitable for or have failed a B-cell receptor pathway inhibitor. Venclyxto monotherapy is indicated for the treatment of CLL in the absence of 17p deletion or TP53 mutation in adult patients who have failed both chemoimmunotherapy and a B-cell receptor pathway inhibitor(92). Approval is based on phase 1 escalation study (93) and in a phase 2 study(94). Oral alkylating agents with or without corticosteroids. Chlorambucil was used for treatment of CLL over 60 years. Label: LEUKERAN is indicated in the treatment of chronic lymphocytic leukemia(95). Although the role of chlorambucil has considerably dimished, regulators paradoxically still accept drugs for registration on the basis of the phase 3 trials, which use chlorambucil in very low doses as comparator (90, 96, 97), while claiming at the same time that the therapeutic success of such therapies is extremely modest. Thus, low doses are still considered standard therapy, although it was shown that middle and especially high doses have significantly greater effectiveness(98-101). A meta-analysis of six trials of immediate versus deferred therapy with chlorambucil showed no difference in OS at 10 years(70). Purine analogs. Fludarabine is a purine analog, one of a group of chemotherapy drugs known as anti metabolites. These stop cells making and repairing DNA. Cancer cells need to make and repair DNA in order to grow and multiply. Label: Fludarabin (gen) is used in the treatment of B-cell chronic lymphocytic leukemia in patients with sufficient healthy blood cell production. First treatment for CLL with this medicine should only be started in patients with advanced disease having disease related symptoms or evidence of disease progression(102). Approval is based on phase 3 randomized study(96). This drug is also used in combination therapies (see below). Bendamustine. Bendamustine is a cytotoxic agent with bifunctional properties of an alkylator and a purine analog. Label; Bendamustin (gen) is used as monotherapy or in combinations with other drugs for treatment of chronic lymphocytic leukemia in cases when combination chemotherapy containing fludarabine is not appropriate(103). Approval is based randomized phase 3 trial for monotherapy(104) and for combinations in phase 2 trials in previously treated(82) and untreated patients (81). Combination therapy. Fludarabine based combinations include FCR, FCOfa, FR, FC. Fludarabine plus caclophosphamide plus rituximab is proved very effective in those patients who can tolerate the treatment. For this reason, in the last 10 years FCR become a golden standard of CIT for fit patients(79). Long term results confirmed overall efficacy and a subset of long-term responers defined by genomic risk groups emerged (105-107).Athough no head to head comparisons are not yet completed in line with new paradigm, the indication is narrowed to a subset of fit patients with hyper mutated IVGH in whom likelihood for very long remission may outweigh concerns of chemotherapy toxicity. Bendamustine combinations are used in those patients in whom fludarabine can not be tolerated. In head to head comparison to FCR, BR combination was found inferior(108). Combination therapy without anti-CD20 monoclnal antibodies. For FC, CVP and CHOP a meta-analysis of ten trials compared combination chemotherapy (before the availability of rituximab) with chlorambucil alone and showed no difference in OS at 5 years(70). Combination with novel agents. It is likely that combinations among BRI and Bcl2 inhibitors with anti-CD20 antibodies will be the base in era of the emerging new paradigm. At presnt only ibrutinib + bendamustin + riruximab and idelalisib + anti-CD20 monoclonal antibodies are approved. Bone marrow and peripheral stem cell transplantations. Although this modality is considered the only option for cure, it is still under clinical evaluation especially in the context of novel agents and new emering paradigm (109, 110). The overall therapeutic effect is a consequence of total therapeutic interventions, including antineoplastic and supportive measures that are particularly important. Criteria for evaluation of response to therapyTable 5 shows the criteria for assessment of therapeutic effect. The criteria generally used the same grounds, based on the estimation of parameters of the tumor mass in different compatrments on the one hand, and the parameters for the assessment of myelopoiesis on the other hand. The criteria are somewhat different in the current NCI / iwCLL criteria (6, 13) and the criteria described below (IGCI, EORTC) (98-100, 111, 112) To monitor the dynamics of the disease (both progression and responsel to therapy) TTM score (described in the clinical stage) is very convenient, because is the only clinical system that is based on continuous, quantitative parameter easy to apply, validated in thousands of patients in various international clinical trials. To estimate the doubling time (DT) is more reliable than just the number of lymphocytes, because it can compensate for changes in the distribution of the occurrence of the tumor mass as after administration of corticosteroids or TKIs, when it may be an increase in the number of leukocytes while reducing nodes or spleen. For this reason NCI/iwCLL critera were recently amended, but are still suboptimal for monitoring of the diesase response(23, 113), while TTM scoring system is much better measuring redistribution od clonal cells among compartments(30, 114). When assessing the response to therapy the complete remission (CR) is assessed equally in TTM and NCI based criteria, but TTM shows the advantage in assessing partial remission (PR) by comparing the total tumor mass before and after treatment, so it is possible to set a minimum threshold for minimal remission (MR), eg reduction of > 25%, partial remission (PR) > 50%, very good PR > 75% and more. Likewise, TTM based criteria are more accurate and without bias estimate the stable disease (SD) and progressive disease (PD). Continuous quantitative character of TTM size allows comparison of trends between the group criterion A (TTM) and group B (function residual normal hematopoiesis). It is possible to evaluate the beneficial antineoplatic effect of the therapy independently of toxic effect on hematopoiesis. Minimal residual disease (MRD) is an important end point, and should reflect no measurable disease in the body. However, MRD testing should be standardized and sample source well defined. By doing this we should avoid reports of MRD negative patients with persistent significant organomegaly. Those cases in fact have clean blood but sometimes may not fulfil common criteria for partial remission, and the term MRD without specification is misleading. Antineoplastic therapy current treatment optionsThere are several treatment options. Therapeutic recommendations summary for first-line treatment and for salvage treatment in major therapeutic stratification groups are shown on respective tables. Management of patients with no accepted criteria for therapy If the patient is not showing any signs of active / progressive / symptomatic disease, the antineoplastic therapy is not recommended, but the patient is monitored and reviewed without therapy. Thus Watch and Wait should be transformed to Watch and Investigate W&I) This view is based on evidence collected in randomized trials during the 80-ies of the last century, when it was shown that chlorambucil based treatment does not contribute to longer survival, moreover, despite the relative ease of controlling symptoms and achieving clinical remission, overall survival was marginally worse(98, 111, 115). Until now there are no data indicating that harm is done by deferring therapy in asymptomatic, stable disease. However, trials that are under way that include novel agents and/or combinations may change this concept, but it will take time since those trials require prolonged follow-up. While for these patients antineoplastic medication is not recommended, standard care should include infection prophylaxis such as annual flu and pneumococcal every 5 years, and in case of infection early treatment. Initial treatment (first-line treatment) The first-line treatment relates to previously untreated patients. All patients in standard care must have clinical indication for treatment initiation (i.e must fulfil criteria stipulated in section 2.5). Recommendation depends on on the risk associated with B-CLL (High or Low) and patient general condition (Fit or Unfit). (Table 6. and Figure 1.) Each stratum will be discussed separately. Initial treatment for patients with no del(17p/TP53 mutation that are fit. (LOW RISK+FIT) As a rule the patients in this stratum (about a third of first line treatment) are younger than 65 years. The therapeutic goal is to be set high, to achieve complete, durable remission and prolong survival and perhaps even offer a possible cure. FCR (fludarabine, cyclophosphamide, rituximab) is recommended as a standard initial therapy for previously untreated fit patients outside clinical trials(79). According to the DCLLSG CLL8 protocol, 6 cycles at intervals of 28 days if the patients tolerate the treatment well, and after EOT, no further treatment is anticipated, only follow-up visits. Long term follow-up identified a subset of patients in whom long and durable response were achieved(106, 107, 116). Those patients had mutated IGHV, no 11q and are less < 65. Other patients may respond, but they tend to relapse soon. For this reason, for fit patients, <65, mutated IGHV and no 11q (nor del(17p)/TP mutation) who fulfill citeria for treatment, FCR is treatment of choice. In others, according to new paradigm it is suggested to avoid chemotherapy, especially FC. In patients who are unsuitable for fludarabine therapy bendamustin + rituximab (BR) can be tried. It seems to be both less toxic and less effective. Ibrutinib is also approved for this indication According to the new paradigm BRI may replace CIT, but at present there is no data of randomized trials to support it. Head to head comparison of FCR or BR with ibrutinib are under way and are likely to reslove this question. Initial treatment for patients with no del(17p)/TP53 abnormality that are unfit. (LOW RISK+UNFIT) Majority of patients (more than 60% of treated in first line) belong to that group. As a rule, the patients are older than 65 years, with commorbidity therefore not capable to tolerate aggressive CIT therapeutic approach, and therefore it's necessary to modify the therapeutic goal and choose remission or stabilization of disease with a well-preserved quality of life. We recommend as a standard chlorambucil plus one of the anti-CD20 antibody(83, 84, 87, 117). Best results are published with chlorambucil + obinutuzumab. Also BR is an option for chemo-immunotherapy for patients with appropriate fitness. Ibrutinib is also approved for this indication beacause of excellent results in this patient subset. According to the new paradigm BRI may replace CIT, but at present there is no direct data to support it. Head to head comparison of ibrutinib+obinutuzumab with chlorambucil+obinotuzumab is under way and is likely to reslove this question. Initial treatment for fit/unfit patients with del(17p)/TP53 mutation. (HIGH RISK+ FIT/UNFIT) In this stratum we expect less than 7% of all patients treated in first line. CIT is contraindicated in this subset, since the TP53 mutated clone is not responding and CIT may enhance unfavorable clonal selection and is therefore harmful. We recommend for both fit and unfit the induction with ibrutinib or idelalisib plus rituximab. Ibrutinib appears superior to idelalisib in all settings as first choice BRI(118-120). In selected cases this therapy can be followed by elective AlloSCT. HDMP plus rituximab(121, 122) or Alemtuzumab(123) should be used if BCI therapy is unavailble. Treatment for relapsed / refractory CLL This relates to previously treated patients. Again, they should fulfil citeria for re-treatment, essentially the same as described in the section 2.5. For this reason, time to progression (measured as PFS) is distinct from time to next treatment (TTNT). The situation in this setting is much more complex, since in addition to four major therapeutic strata special attention should be paid to previous treatment(s) (type of treatment, number of treatment lines and the period that had lapsed from previous treatment etc.). In principle, with the exception of very late relapses, the patients require more therapy to achieve less response. Since the vast majority (>90%) of all CLL first-line treated patients have no TP53 abnormality, their treatment allocation was essentially dependent on their general condition (Fit or Unfit). Thus, the fit patients received more agressive treatment aiming MRD negativity (hopefully the erradication of the disease) while the unfit patients receive less agresive treatment that is less likely to achieve MDR negativity and consequently the therapeutic aim is less ambitious. The relapse is, therefore, primarily linked to the first-line therapeutic stratum. It is an indicator of respective therapy failure. In principle, the longer the period to relapse the more effective first-line treatment was. General principles of therapeutic strategies in relapsing / refractory patients are shown on Table 7. and Figure 1. The relapsing patient in early relapse is considered refractory and requires change of therapy. If the treatment result in remission of long duration, it is reasonable to try in relapse again the same treatment that has proved effective For this reason, relapses will be described as a function of first-line stratification therapeutic failures. However, in all patients in relapse TP53 status should be checked (before each new line of therapy) to assess wheather the risk grade has changed in comparison to front-line stratification. Most of the patients currently relapsing early were in first line treated by CIT adjusted by fitness status. Occasionally, some patients in first relapse were in first line treated by chemotherapy (chlorambucil, fluradabine alone or in combination with cyclophosphamide). Patients relapsing from the LOW RISK+FIT stratum Patients relapsing early are considered refractory, and shoud be treated with ibrutinib, or idelalisib plus rituximab. If the anti BCR drugs cannot be provided, the current options include BR, HDMP+R, FA, other chemo-immuno therapy, ofatumumab and alloSCT. Patients relapsing late who have not acquired a TP53 abnormality, remain fit enough for fludarabine-based treatment and in whom there is a clinical indication for treatment, may receive FCR, provided that they have mutated IGHV, no TP53 nor 11q (109, 124). If the patient at the time of relapse changes to unfit stratum, the relapse treatment described in respective section applies. Patients relapsing from the LOW RISK+UNFIT stratum Over 50% of all treated patients belong to this group. In this group less agressive treatment was applied in front-line due to the fact that those patients are not likely to tolerate FCR. Obtained respone is less likely MRD negative and relapses are expected in the wide range from early (less then 24 months) or late (more then 24 months). In case of early relapse patient is considered refractory to given treatment and ibrutinib or idelalisib + rituximab is reccommended. Less benefit can be expected from BR or chlorambucil+antiCD20 or HDMP + R. In selected responsive cases ofatumumab maintenance. In case of a late relapse the patients relapsing after chlorambucil can be retreated with chlorambucil + anti-CD20 monoclonal antibody(117). The therapy may be repeated several times (in function of achieved duration) until the duration of remission is not shortened to 2 years, after which it is justified to go to the second line therapy (ibrutinib or idelalisib+R). Patients relapsing from the HIGH RISK+FIT/UNFIT stratum Those patients are at particularly high risk. If the patients did not receive BRI treatment, those drugs are reccommended. If the patients relapse on BRI drugs, venetoclax or alternative BRI is indicated. If those are not available HDMP + R or Alemtuzumab ± R can be tried. If the fit patients are not previousely allo tranplanted, a reinduciton should be considered with different combination (including ofatumumab) and if succesful proceed to transplantation. In selected responsive cases ofatumumab maintenance may be considered(125). Consolidation / Maintenance therapy The observation that an MRD negative remission is associated with prolonged progression free survival both in previously untreated(126) and relapsed(127) patients. This has lead to studies of additional treatment in patients with residual disease post therapy. In line with new paradigm although chlorambucil maintenance can prolong survival(128) maintenance treatment should be chemotherapy free. Ofatumumab in selective responsive patients was shown to prolong PFS, but nor OS(129) and is approved for this indication by FDA. Lenalidomide was also shown to improve PFS, but is not yet approved by regulators(130, 131). As described earlier, BRI therapies are given for a prolong period of time to maintain the response as distinct from chemo-immunotherapy. Early (months) period of treatment is characterized by marked tumor mass redistribution so that the monitoring should be adjusted accordingly (see section for response criteria). The role of allogeneic transplantation The indication for allogeneic SCT is currently changing in line with exellent results of BRI and venetoclax. Current indications include poor responders to BRI and Bcl-2 antagonist, and appearance of Richter syndrome(110). Radiotherapy can provide effective palliation in cases with symptomatic bulky lymphadenopathy and should be offered to patients for whom chemo-immunotherapy has been ineffective or is contra-indicated. Low doses of external beam radiotherapy (2 x 2Gy) can be highly effective in this situation and a higher dose (30 Gy in 2-3 Gy fractions) may be required in patients with transformed aggressive disease or those known to have a TP53 abnormality(132). The biological similarities between SLL and CLL are so close that a similar response to treatment could be expected. This is supported by MDACC single centre retrospective study(133). Indications for, and choices of treatment are the same as for CLL. Rare patients in whom SLL is diagnosed following biopsy of an enlarged lymph node in the absence of detectable disease at any other site, may be offered local radiotherapy with curative intent. Risk of other diesaesInfectionsRisk of infections is related to progressive defect both in antibody- and cell-mediated immunity. In addition, the therapy may worsen immune impairment, particularly fludarabine and anti-CD20 antibodies, but also the B-cell receptor inhibitors. For this reason, infections represent the frequent cause of morbidity and mortality in CLL. Infections are typically bacterial and should be treated early. For those who have repeted infection and require repeated antibiotics imunoglobulin relacement therapy should be considered. Prophlyllactic vaccination is advised, but llive vaccine should be avoided. Autoimmune complicationsAutoimmune complications are common in CLL occurring in 10-20% of patients.(134) These almost exclusively target blood cells, most commonly red blood cells. The haemolytic anaemia (AIHA) is predominant and immune thrombocytopenia (ITP) is 4-5 timesless common. A bone marrow aspirate is usually required to confirm the diagnosis of autoimmune cytopenia. AIHA or ITP should be treated before deciding whether therapy for CLL is needed. First line therapy is prednisolone. Second line therapies for patients intolerant of or refractory to steroids, include cyclosporine, intravenous immunoglobulin (ITP), thrombopoietin mimetic agents (ITP), CVP, low-dose cyclophosphamide, rituximab, alemtuzumab and splenectomy(135). CLL treatment may be initiated to control recurrent or refractory AIHA/ITP. Rituximab containing regimens are recommended in patients who do not have a TP53 abnormality. If AIHA/ITP develops during CLL treatment the same regimen should only be used again in that patient with extreme caution and if no effective alternative is available. Autoimmune neutropenia usually responds to GCSF. Prolymphocytic transformationB-cell prolymphocytic transformation is diagnosed in progressive organomegaly and lymphocytosis with increase of prolymphocytes >55% and is treated as agressive CLL. It occurs rarely in <1% of cases. Richter syndromeRichter syndrome is CLL transformation to agressive lymphoma usually DLBCL or Hodgkin like. LN biopsy is mandatory for diagnosis, PET may be helpful. It occurs in about 2-7% of patients. While novel agents are under investigation, CIT is still recomended approach. Depending on the histological sub type of lymphomatous transformation, patients who are suitable for intensive therapy should receive regimens currently employed for either primary diffuse large B cell lymphoma (Richter syndrome) or Hodgkins lymphoma. Younger patients who achieve a good response are candidates for allogeneic stem cell transplantation. Check point inhibitor pembrolizumab was shown activity in RS, but not in CLL(136-138). Myelodisplastic syndrome and acute leukemiaAlthough MDS and AML are uncommon in CLL, the rate of therapy related to fluadabine based CIT is about 5% and much higher after autologous stem cell transplantation. Response to therapy is poor. Wheather novel agents that do not induce genotoxic stress to stem cells reduce the incidence of this serious complication is currenty under evaluation.
Supportive therapySupportive therapy plays an important role in management, and is carried out in accordance with generally accepted good clinical practice(16, 17). Table 8 shows the basic characteristics of supportive therapy in B-CLL. This covers the area of vaccination, anti-infective prophylaxis, respiratory recurrent infections requiring IV antibiotics and hospitalization, immunoglobulin replacement therapy, blood transfusions, and tumor lysis.
Extended version and updates can be found on KROHEM website: http://www.krohem.hr/Documents/AMENDMENT%20KROHEM%20CLL%20GUIDELINES%20v1.%202016%20%20ENGL.pdf
Table 1 Steps and aims in diagnostic process that lead to definition of therapeutic goal and strategy
Comment: table shows diagnostic steps. Steps 1-4 are made in single visit. Each step is different with respect to aim, decision criteria as well as the extent of work-up. Last column describes proposed classification categories. Only about 10% of paients have indication for therapy at diagnosis. Others are followed-up repeatedly until criteria for therapy are reached (step 5). The scope of work-up is different in each step, pre-treatment evaluation being the most complete aiming to provide all of the necessary elements for patients stratification and definition of therapeutic goal. The overall goal of diagnostic process is to enable individualization of therapy, definition of therapeutic aim and strategy, by implementing general principles on individual case.
Table 2. Clinical stages and TTM
Table 3 Criteria for active (progressive / symptomatic) disease
Hypogammaglobulinemia, monoclonal or oligoclonal paraproteinemia, or absolute lymphocyte count, do not in themselves constitute an indication for therapy. It is out of 8 groups of criteria theoretically possible to identify 11 individual indications based on exceeding a threshold, 3 dynamic evaluation of continuous quantitative parameters, where individual trends can be compared and thereby gain additional derived criteria, and 4 qualitative assessments. Although in principle sufficient presence of at least one indication, we should avoid making decisions on an isolated indication. It is clear that a larger number of indications further reinforces the decision to begin treatment. It is possible to decide that the patient needs to document the presence of at least two or more of the above indications for active (progressive / symptomatic) disease. The indication for treatment (according to KROHEM guidance) should be documented in patient records!
Table 4 General schedule of investigations before, during and after therapy
* Monitoring protocol frequency varies depending on the clinical condition from several weeks to several months, or even one year if the situation is stable, without change. However, in the emergence of new circumstances, it is necessary to check- up early. **these repetitive visits depend on treatment applied. For novel agents with at present unlimited duration, this visits shoud enable detection of treatment failure; bpreferred, but not required tests;
Table 5 Definition of response to treatment (NCI updated guidelines, Blood 2008)
Group A criteria define tumor mass, group B criteria define hematopoetic system (or bone marrow) function. 1 CR (complete remission): all criteria must be present, and patients must be without general symptoms associated with CLL; PR (partial remission): at least two criteria in group A plus one in group B must be present; SD (stable disease) is the absence of progressive disease (PD) if at least PR is not reached; PD (progressive disease): at least one criterion from gropus A or B must be present. 2 The sum of the products of multiple lymph nodes (as evaluated by means of CT in clinical trials or physical examination in general practice). 3 These parameters are irrelevant for
certain types of responses. Figure 1 Flow chart describing 2017 paradigm shift
Figure1. Flow chart deccribing B-CLL diagnosis and treatment 2017 paradigm shift. This chart shows diagnostics based decision steps (yellow rectangles) and their sequence (yellow arrows), currently approved therapies by EMA (in2/2017) (blue rectangles), as well as the sequence for first line treatment (green arrows) and salvage treatment lines (blue arrows). Minority of patients (about 10%) at diagnosis present with indication for treatment, while majority are observed until the criteria for treatment are met. This part (framed) did not change. When the indication is present, B-CLL patients are eligible for first line treatment. In this part major changes occurred because of recent approval of new options. Ibrutinib monotherapy is approved as continuous treatment of undetermined duration or until progression or unmanageable toxicity for all patients strata because of favorable efficacy/tolerance ratio in disease control. This is a new approved option so that ibrutinib could be used to start the new path. In case of progression or toxicity, patients qualify for second line treatment (approved option is venetoclax). If this fails, patient is eligible for experimental treatments (combinations of novel agents with immunotherapy, allogeneic stem cell transplantation, CAR-T cell therapy and like). Theoretically, all of this could be done without further diagnostics and stratification, while avoiding chemotherapy. However, head to head comparison data between novel agents and chemo-immunotherapy (CIT) are still lacking. CIT, although associated with higher short and long term toxicities, proved for some patient subsets to be highly effective in achieving long, durable remissions and perhaps even a cure. It was therefore in CIT era essential to identify those who will respond and number of predicting factors emerged in this setting. The NGS has revealed that intratumoral heterogeneity and genomic changes can be used for the better CIT response prediction. Most important for CIT clinical use are two predictors: del(17p)/TP53mut and IGHV mutation status. The first can identiy patients subset in which chemotherapy is ineffective and even contraindicated because of inducing adverse clonal evolution and the second identify disease type, where patients with unmutated IGHV poorly respond to CIT and even if respond, the response is short and clones that are more resistant emerge. Both predictors are considered standard minimum for stratification. If adverse features are present, the patients should be treated with ibrutinib or idelalisib in first line. Others may continue towards CIT that is tailored according to age and comorbidities. Fit patients qualify for FCR, unfit for Clb+Obi or like and patients in between for BR. If they relapse late, the CIT may be repeated, tailored on current fitness, while early relapsed/refractory patients qualify for BRI or venetoclax salvage. At present, baseline stratification based on genetically defined risk, as well as on age and comorbidities to tailor treatment intensity is still needed for CIT, although fitness is currently not important for novel agents. The current, CIT based paradigm (shown horizontally) is losing importance and the new paradigm (shown vertically) is likely to take over. However, it will require identification of new important predictors along the new path, since majority of predictors identified for CIT lose their power in new setting. As data accumulate, the new predictors will emerge for this setting. High throughput NGS has begun to identify new predictors for targeted therapy response as well as new predictors of failure on molecular level, as treatment proceeds. All this may eventually lead to a new upfront stratification for risk adapted precision medicine therapy in B-CLL. The ongoing trials and head to head comparison of novel agents and their combinations with immunotherapy versus CIT are under way. They will hopefully resolve current dilemmas. Novel therapy research including genomic diagnostics is likely to offer new options that will eventually lead to time limited therapies, without chemotherapy. Dg = diagnosis; WW(I) = Watch and Wait (Investigate); Ind
= indications for treatment; PF = predictive factors; Late R = late relapse;
Early R/R = early relapsing or refractory; IBR = ibrutinib; IDELA = idelalisib;
VEN = venetoclax; Exp = experimental treatment; CIT = chemo-immunotherapy; Table 6 FIRST LINE TREATMENT OF CLL (KROHEM v1 2017)
Clinical trials are highly recommended for all subsets, we strongly believe that they improve the level of care. a Projected percentages are based on compiled data from western countries and Croatia. b Percentages of patients with distinct general condition and molecular genetics groups refer to treated patients. Fit patients are less than 65 years of age and with CIRS score less than 6. Younger patients with CIRS score of 6 and more and patients with 65 years or more (regardless of CIRS score) qualify as unfit. c Standard treatments are in order of preference, all are 2A or less according to NCCN consensus, treatments with higher grade are marked (1). d In patients with hipermutated IGHV and no 11q. e for les fit patients. f Alemtuzumab is withdrawn from market, but can be obtained free of charge from producer upon request FCR (fludarabine, cyclophosphamide and rituximab); B = bendamustin; Chl = chlorambucil; R = rituximab; Obi = obinutuzuman; Ofa = ofatumumab; A = alemtuzumab; HDMP (high dose methylprednisolone).
Table 7 TREATMENT OF RELAPSED/REFRACTORY CLL (KROHEM v1 2017)
The guidelines for salvage treatment are more complex than in first line treatment. It should take into consideration additional criteria depending on type of treatment in first line, and on the observed duration of response. Clinical trials are highly recommended for all subsets, we strongly believe that they improve the level of care. a Projected percentages of early and late relapses are based on KB Dubrava data for 2015 and 2016. Percentages of unfit patients and patients with del(17p) tend to increase. Fit patients = less than 65 years of age and with CIRS score less than 6. Younger patients with CIRS score of 6 and more and patients with 65 years or more qualify as unfit; b Standard treatments are in order of preference, but for each individual patients should be based on integration on clinical data and patients preference. All treatments are 2A according to NCCN consensus, treatments with higher grade or lower grade are marked; c in patients who are unsuitable for or have failed a B-cell receptor pathway inhibitor and chemo-immunotherapy; d if not in 1stline e Alemtuzumab is withdrawn from market, but can be obtained free of charge from producer upon request; f in patients who are unsuitable for or have failed a B-cell receptor pathway inhibitor; g ofatumumab is found to significantly prolong PFS in responsive patients in second or third response to chemoimmunotherapy, approved by FDA. FCR (fludarabine, cyclophosphamide and R); B = bendamustin; Chl = chlorambucil; R = rituximab; Obi = obinutuzumab; Ofa = ofatumumab; A = alemtuzumab; Allo SCT = allogeneic stem cell transplantation; HDMP (high dose methylprednisolone); antiCD20 (ofatumumab or obinutuzumab or rituximab). Table 8 Supportive therapy in patients with B-CLL
References
http://www.nature.com/articles/nrdp201696#supplementary-information. 5. PDQ. Chronic Lymphocytic Leukemia Treatment (PDQ(R)): Health Professional Version Bethesda (MD)2017 [cited 2017 2017-2-22]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26389470. 10.3324/haematol.2011.042333. PubMed PMID: 21546505; PubMed Central PMCID: PMC3148908. 9. Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019-32. Epub 2011/02/09. doi: blood-2011-01-293050 [pii] 10.1182/blood-2011-01-293050. PubMed PMID: 21300984; PubMed Central PMCID: PMC3109529. 16. Zelenetz AD, Gordon LI, Wierda WG, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. V2.2017 - 21 February 2017 2017 [2017 Mar 1]. Available from: https://www.nccn.org/professionals/physician_gls/pdf/cll.pdf 18. Surveillance, Epidemiology and End Results (SEER) Program [Internet]. NCI. 2017 [cited 2017 Feb 22]. Available from: http://seer.cancer.gov/statfacts/html/clyl.html. 19. HMNR. Haematological Malignancy Research Network (HMRN). Epidemiology & Cancer Statistics Group at the University of York; 2017 [2017 Feb 22]. Available from: http://www.hmrn.org/statistics/incidence. 22. Novak I, Jaksic O, Kulis T, Batinjan K, A Z. Incidence and mortality trends of leukemia and lymphoma in Croatia, 1988-2009. . Croat Med J. 2012;53(2):115-23. 23. Nieto WG, Almeida J, Romero A, Teodosio C, Lopez A, Henriques AF, et al. Increased frequency (12%) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor flow cytometry approach. Blood; July 2, 20092009. p. 33-7. 27. Binet JL, Auquier A, Dighiero G, Chastang C, Piguet H, Goasguen J, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer. 1981;48(1):198-206. doi: 10.1002/1097-0142(19810701)48:1<198::aid-cncr2820480131>3.0.co;2-v. 42. Bergmann M, Eichhorst B, Busch R, et al. Prospective evalution of prognostic parameters in early stage chronic lymphocytic leukemia (CLL): results of the CLL1-protocol of the German CLL Study Group (GCLLSG) [abstract]. Blood. 2007;110:625. 43. Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003;348(18):1764-75. Epub 2003/05/02. doi: 10.1056/NEJMoa023143 348/18/1764 [pii]. PubMed PMID: 12724482. 48. Arruga F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014;28(5):1060-70. doi: 10.1038/leu.2013.319. PubMed PMID: 24170027. 53. Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, et al. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2008;23(1):117-24. doi: http://www.nature.com/leu/journal/v23/n1/suppinfo/leu2008274s1.html. 54. Hallek M, Wanders L, Ostwald M, Busch R, Senekowitsch R, Stern S, et al. Serum beta(2)-microglobulin and serum thymidine kinase are independent predictors of progression-free survival in chronic lymphocytic leukemia and immunocytoma. Leuk Lymphoma. 1996;22(5-6):439-47. doi: 10.3109/10428199609054782. PubMed PMID: 8882957. 66. Pflug N, Bahlo J, Shanafelt TD, Eichhorst BF, Bergmann MA, Elter T, et al. Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood. 2014;124(1):49-62. doi: 10.1182/blood-2014-02-556399. PubMed PMID: 24797299; PubMed Central PMCID: PMCPMC4260976. 10.1182/blood-2009-08-207126. PubMed PMID: 19850738; PubMed Central PMCID: PMC2941409. 71. EMA. Imbruvica: EPAR - summary for the public [updated 2016-10-6; cited 2017 2017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/003791/WC500177778.pdf. 75. EMA. Zydelig: EPAR - summary for the public: EMA; [updated 2016-11-142017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/003843/WC500175380.pdf. 78. EMA. Mabthera: EPAR - summary for the public: EMA; [updated 2016-8-2; cited 2017 2017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000165/WC500025815.pdf. 10.1200/JCO.2009.26.4556. PubMed PMID: 20194844. 81. Fischer K, Cramer P, Busch R, Bottcher S, Bahlo J, Schubert J, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2012;30(26):3209-16. Epub 2012/08/08. doi: 10.1200/JCO.2011.39.2688 JCO.2011.39.2688 [pii]. PubMed PMID: 22869884. 82. Fischer K, Cramer P, Busch R, Stilgenbauer S, Bahlo J, Schweighofer CD, et al. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2011;29(26):3559-66. doi: 10.1200/JCO.2010.33.8061. PubMed PMID: 21844497. 85. EMA. Gazyvaro: EPAR - summary for the public [2017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002799/WC500171597.pdf. 86. EMA. Arzerra: EPAR - summary for the public [updated 2017-2-82017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/001131/WC500093092.pdf. 88. EMA. MABCAMPATH: EPAR - summary for the public [updated 2012-8-152017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000353/WC500025261.pdf. 10.1016/S1470-2045(11)70242-X. PubMed PMID: 21992852. 10.1200/JCO.2007.12.9098. PubMed PMID: 17984186. 10.1200/JCO.2011.35.9695. PubMed PMID: 22493413. 92. EMA. Venclyxto: EPAR - summary for the public [updated 2016-12-212017-2-22]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/004106/WC500218803.pdf. 95. FDA. Leukeran [updated 20032017-2-22]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2003/10669slr028_leukeran_lbl.pdf. 96. Rai KR, Peterson BL, Appelbaum FR, Kolitz J, Elias L, Shepherd L, et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med. 2000;343(24):1750-7. doi: 10.1056/NEJM200012143432402. PubMed PMID: 11114313. 102. HALMED. Fludarabin [updated 2013-10-312017-2-22]. Available from: http://www.halmed.hr/en/Lijekovi/Baza-lijekova/Fludarabin-Pliva-25-mgml-koncentrat-za-otopinu-za-injekciju-ili-infuziju/10171/. 103. HALMED. Bendamustin [updated 2016-1-212017-2-22]. Available from: http://www.halmed.hr/en/Lijekovi/Baza-lijekova/Bendamustin-PharmaS-25-mgml-prascaronak-za-koncentrat-za-otopinu-za-infuziju/12264/. 104. Knauf WU, Lissichkov T, Aldaoud A, Liberati A, Loscertales J, Herbrecht R, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol. 2009;27(26):4378-84. doi: 10.1200/JCO.2008.20.8389. PubMed PMID: 19652068. 110. Dreger P, Schetelig J, Andersen N, Corradini P, van Gelder M, Gribben J, et al. Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents? Blood. 2014;124(26):3841-9. doi: 10.1182/blood-2014-07-586826. PubMed PMID: 25301705; PubMed Central PMCID: PMCPMC4276025. 10.1200/JCO.2012.43.3748. PubMed PMID: 22778323; PubMed Central PMCID: PMC3410400. 114. Jaksic O, Kinda SB, Aurer I, Carzavec D, Rogulj IM, Haris V, et al. REDISTRIBUTION PATTERN AND ASSESSMENT OF EARLY RESPONSE TO IBRUTINIB IN CLL BY TOTAL TUMOR MASS SCORE (TTM) - KROHEM CLL2 STUDY. Haematologica. 2016;101:723-. PubMed PMID: WOS:000379484602583. 120. Mato AR, Hill BT, Lamanna N, Barr PM, Ujjani CS, Brander DM, et al. Optimal Sequencing of Ibrutinib, Idelalisib, and Venetoclax in CLL: Results from a Large Multi-Center Study of 683 US-Patients: ASH; 2016 [2017-2-22]. Available from: https://ash.confex.com/ash/2016/webprogram/Paper93270.html. 10.1182/blood-2011-03-341032. PubMed PMID: 21670470. 125. Fiegl M, Falkner F, Steurer M, Zojer N, Hopfinger G, Haslbauer F, et al. Successful alemtuzumab retreatment in progressive B-cell chronic lymphocytic leukemia: a multicenter survey in 30 patients. Ann Hematol. 2011;90(9):1083-91. Epub 2011/02/26. doi: 10.1007/s00277-011-1192-5. PubMed PMID: 21350830. 126. Bosch F, Ferrer A, Villamor N, Gonzalez M, Briones J, Gonzalez-Barca E, et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: high response rate and disease eradication. Clin Cancer Res. 2008;14(1):155-61. Epub 2008/01/04. doi: 14/1/155 [pii] 10.1158/1078-0432.CCR-07-1371. PubMed PMID: 18172266. 10.1200/JCO.2005.04.021. PubMed PMID: 15738539. 128. Jaksic B, Brugiatelli M, Stelitano C, Morabito F, Callea V, Jaksic O, et al. Chlorambucil maintenance significantly improves survival in B-cell chronic lymphocytic leukemia (CLL): Meta-analysis of IGCI CLL trials. Blood. 1997;90(10):2362-. PubMed PMID: WOS:A1997YG42402358. 130. Fink AM, Bahlo J, Robrecht S, Al-Sawaf O, Aldaoud A, Hebart H, et al. Lenalidomide Maintenance after Front Line Therapy Substantially Prolongs Progression Free Survival in High Risk CLL: Interim Results of a Phase 3 Study (CLL M1 study of the German CLL Study Group 2016 [2017-3-1]. Available from: https://ash.confex.com/ash/2016/webprogram/Paper89160.html. 131. Results of the Phase 3 Study of Lenalidomide Versus Placebo As Maintenance Therapy Following Second-Line Treatment for Patients with Chronic Lymphocytic Leukemia (the CONTINUUM Trial) [Internet]. 2016 [cited 2017-3-1]. Available from: https://ash.confex.com/ash/2016/webprogram/Paper93104.html. 10.1016/j.radonc.2011.05.013. PubMed PMID: 21664710. 10.1200/JCO.2006.09.4508. PubMed PMID: 17925562. 10.3324/haematol.2010.036152. PubMed PMID: 21242190; PubMed Central PMCID: PMC3084923. 136. Brusa D, Serra S, Coscia M, Rossi D, D'Arena G, Laurenti L, et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica. 2013;98(6):953-63. doi: 10.3324/haematol.2012.077537. PubMed PMID: 23300177; PubMed Central PMCID: PMCPMC3669453. 138. Ding W, Le Rademacher J, al e. Paper: PD-1 Blockade with Pembrolizumab in Relapsed CLL Including Richter⼯span>€s Transformation: An Updated Report from a Phase 2 Trial (MC1485) 2016. Available from: https://ash.confex.com/ash/2016/webprogram/Paper95426.html. | ||||||||